


















Explore Our Degree Programs



Recent Research Highlights

Microplastics
Prof. Cizdziel recently examined the occurrence, transport, and fate of microplastics in a waste stabilization pond. MORE…
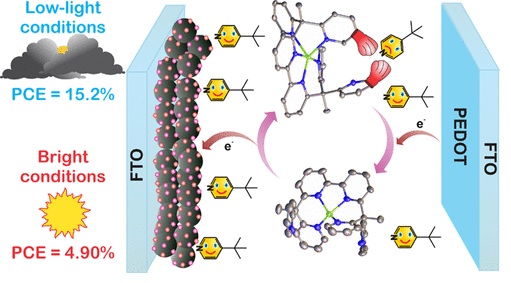
Low Light Solar Cells
Prof. Delcamp demonstrated a new Cu redox shuttle that is an intriguing candidate for implementation with narrow MORE…
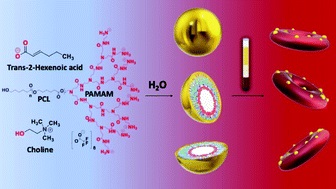
Nanoscale Copolymer Formulations
Prof. Tanner showed that by controlling the physical chemistry of polymer-IL interactions and assembly on the nanoscale MORE…


